炎性肌病是一组以免疫系统攻击自身骨骼肌为核心的自身免疫性疾病,临床表现复杂、亚型繁多、易漏诊误诊。近日《新英格兰医学杂志》(NEJM)发表综述,全面梳理了炎性肌病的现代分类、临床特征、自身抗体谱、病理机制、预后评估与靶向治疗进展。本文结合临床实践,将这篇顶级综述转化为易懂、实用的临床指南,助力风湿科、神经内科、皮肤科医师快速掌握核心要点。

传统分类中“多发性肌炎/皮肌炎”的概念已被全新分型体系取代。目前国际公认将特发性炎性肌病分为五大亚型,各亚型在临床表现、受累部位、预后和治疗上差异极大:
1. 包涵体肌炎(IBM)
2. 免疫介导性坏死性肌病(IMNM)
3. 抗合成酶综合征(ASS)
4. 重叠性肌炎(OM)
5. 皮肌炎(DM)
简单划分:IBM与IMNM以肌肉受累为主,预后看功能;ASS、OM、DM为系统性疾病,可累及皮肤、肺、关节,严重可致命。
过去常说的“多发性肌炎”,如今已被证实绝大多数属于其他亚型,真正意义上的“多发性肌炎”已非常罕见。
1、发病率:0.79/10万人年,患病率14/10万。
2、性别:女性高发,唯独IBM男性更多。
3、年龄:皮肌炎可发生于任何年龄(幼年型<15岁);IBM几乎均>35岁,高峰约60岁;IMNM、ASS、儿童罕见。
4、遗传与环境:HLA区域为易感基因;环境诱因包括感染、吸烟、紫外线、肿瘤、他汀类药物等。
(一)肌肉受累:强弱、快慢、分布完全不同
1. 包涵体肌炎(IBM)起病慢、隐匿、不对称;
经典表现:股四头肌无力+手指屈肌无力,几乎具有病理特征性;
肌酸激酶(CK):正常至中度升高;
MRI:肌肉炎症+脂肪替代并存。
2. 免疫介导性坏死性肌病(IMNM)
起病急、进展快;
主要累及腰盆部近端肌;
CK:显著升高(20~30倍);
治疗延迟会出现广泛肌纤维坏死与脂肪替代。
3. 抗合成酶综合征(ASS)
对称性近端肌无力,亚急性起病;
CK明显升高,但肌无力程度多为中度;
约1/3患者无肌炎表现,仅以肺、关节、皮肤受累起病。
4. 皮肌炎(DM)
1/3患者无肌无力或极轻(无肌病性皮肌炎);
对称性无力,三角肌常最先受累;
CK可正常,与肌无力程度不相关。
(二)肌外表现:系统性受累决定生死
1. 皮肌炎:皮肤损害为标志——向阳疹、Gottron丘疹/征、V形征、披肩征、甲周病变;8%无皮疹(抗NXP2型)。
2. 抗合成酶综合征:技工手、雷诺现象、多关节炎、间质性肺病(ILD)。
3. 共同高危点:ASS与抗MDA5型DM的ILD发生率>80%,是主要致死原因。
4. IBM与IMNM:基本无明显肌外表现。
1. 自身抗体:炎性肌病的“分子指纹”
约70%患者可检出自身抗体,肌炎特异性抗体(MSA)直接决定分型、预后与肿瘤风险,是现代诊断的核心。
(1)各亚型对应抗体速记
IBM:抗cN1A(敏感性高、特异性有限);
IMNM:抗SRP、抗HMGCR(他汀相关多见);
ASS:抗Jo-1最常见,还有PL-7、PL-12、EJ、OJ等;
DM:抗Mi-2、TIF-1γ、NXP2、MDA5、SAE;
重叠肌炎:抗PM/Scl、Ku、RNP 。
(2)肿瘤风险分层(临床必记)
高风险:DM血清阴性(>40岁)、抗TIF-1γ、抗NXP2、抗OJ ;
中风险:抗Mi-2、MDA5、SAE、抗HMGCR;
低风险:IBM、ASS多数抗体、IMNM抗SRP、重叠肌炎。
2. 病理检查:诊断的“最终裁判”
肌肉活检:金标准,尤其适用于抗体阴性病例、IBM确诊;
皮肌炎:束周萎缩、血管病变、C5b-9沉积;
合成酶综合征:束周肌纤维坏死、肌束膜炎;
IMNM:广泛肌纤维坏死、炎症轻;
IBM:镶边空泡、CD8+T细胞浸润.
3. 鉴别诊断:别把这些病当成炎性肌病
遗传性肌病(肢带型肌营养不良、Dysferlin肌病);
药物性肌病(他汀、糖皮质激素);
甲状腺功能减退;
感染性肌炎(HIV、旋毛虫、钩体);
代谢性肌病。
关键提示:激素治疗无效→高度警惕遗传肌病或IBM。
表1: 肌炎特异性自身抗体与肌炎相关性自身抗体特征
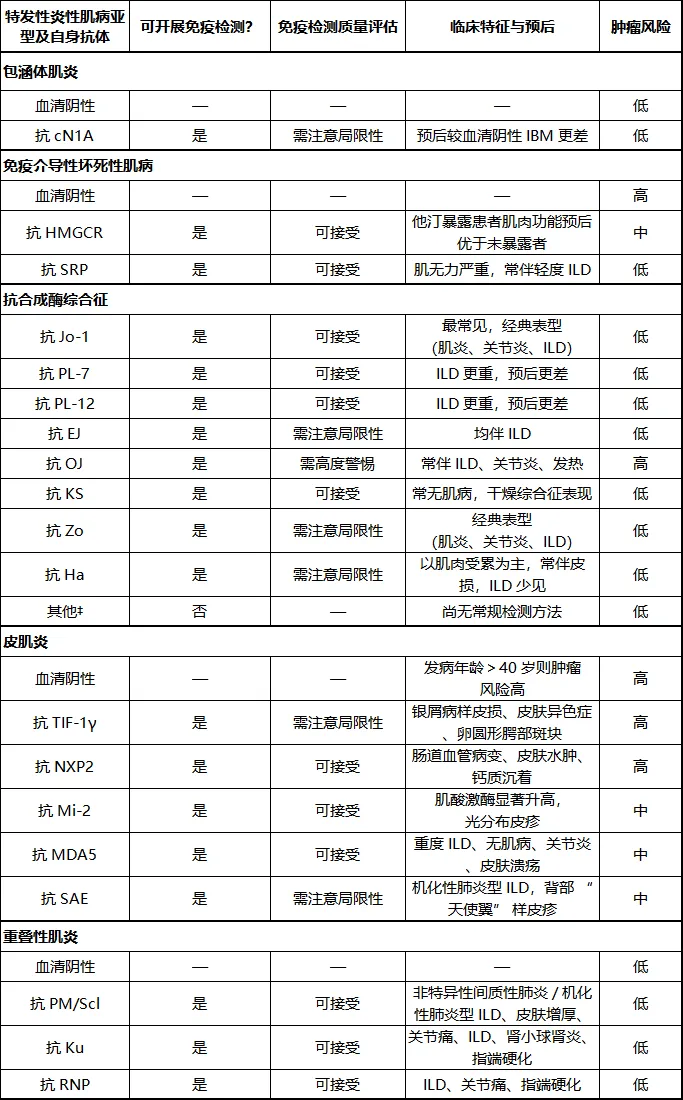
1. 生存预后
确诊1年死亡率约10%,死因主要为肿瘤+肺部并发症;
10年生存率:ASS抗Jo-1型约70%,非Jo-1型仅47%;
抗MDA5型DM:快速进展ILD发生率30%,死亡率>50%;
肿瘤关联:约10%患者在确诊前后3年内发生恶性肿瘤。
2. 肌肉功能预后
不可逆损伤:肌纤维被脂肪/纤维组织替代;
最重:IMNM、IBM(IBM通常10年内需行走辅助);
较好:DM、ASS(规范治疗后功能显著改善);
关节受累:ASS与重叠肌炎多为非破坏性关节炎。
1. IBM:炎症+变性双重机制,CD8+T细胞介导细胞毒,伴随蛋白聚集、自噬障碍、线粒体异常。
2. IMNM:抗体直接致病,抗HMGCR/SRP抗体损伤肌纤维,补体激活,B细胞关键作用;
3. ASS:抗tRNA合成酶抗体驱动,肺与肌肉同时受累,体液免疫核心。
4. DM:Ⅰ型干扰素病,干扰素-β导致内皮与肌纤维损伤,属于“获得性干扰素病”。
1. 包涵体肌炎(IBM):治疗最棘手
传统免疫抑制剂无效:激素、IVIG、甲氨蝶呤均不改善肌力;
大样本试验:阿里莫克隆、比马鲁单抗均失败;
在研新药:西罗莫司、抗KLRG1单抗(尚在临床试验);
唯一推荐:规律康复训练,延缓功能衰退。
2. 免疫介导性坏死性肌病(IMNM)
一线:大剂量激素+甲氨蝶呤;
抗HMGCR型:联合IVIG;
抗SRP型:利妥昔单抗获益更显著;
难治病例:达雷妥尤单抗、CD19 CAR-T细胞治疗(个案成功)。
3. 抗合成酶综合征(ASS)
B细胞耗竭为核心:利妥昔单抗疗效确切;
合并ILD:吗替麦考酚酯、环磷酰胺、钙调磷酸酶抑制剂;
重症ILD:三联免疫抑制+抗纤维化(尼达尼布)。
4. 皮肌炎(DM)
一线:激素+甲氨蝶呤(优于单用激素);
首选靶向:IVIG(唯一获批标准治疗);
重症/难治:JAK抑制剂、TYK2抑制剂、抗Ⅰ型干扰素受体单抗;
抗MDA5型危重症:激素+环磷酰胺+钙调磷酸酶抑制剂三联,必要时行肺移植。
5. 通用原则
所有亚型均推荐:有氧+抗阻训练(每周3次),安全且改善生活质量;
肿瘤筛查:按抗体风险分层,高风险者需全面筛查;
肺部评估:基线及定期胸部高分辨CT。
1. 抗体清除:FcRn阻断剂、达雷妥尤单抗、CAR-T 。
2. 抗纤维化:尼达尼布用于肌炎相关ILD。
3. 干扰素通路靶向:JAK/TYK2抑制剂、抗IFN-β/受体单抗。
4. 细胞毒性T细胞靶向:用于IBM的新型免疫调节。
1. 炎性肌病分5型:IBM、IMNM、ASS、重叠肌炎、DM。
2. 抗体定分型、定预后、定肿瘤风险。
3. IBM:手指屈肌+股四头肌受累,激素无效。
4. IMNM:CK极高、快速坏死,抗体介导。
5. ASS:技工手+关节炎+ILD,利妥昔单抗有效。
6. DM:皮疹+肌无力,MDA5型肺凶险,JAK抑制剂有效。
7. 治疗必须分型施治,拒绝“一刀切”。
8. 康复训练是所有亚型的基础治疗。
炎性肌病虽为罕见病,但误诊率高、危害大。随着自身抗体检测普及与靶向药物问世,本病已从“难治性”走向“可精准治疗”。临床医师应建立“抗体优先、分型诊治”的现代理念,尽早识别高危病例,改善患者长期预后。
参考文献:ALLENBACH Y, BENVENISTE O. Inflammatory Myopathies[J]. N Engl J Med, 2026, 394(19): 1925-1938.